UniProt is a comprehensive, high-quality and freely accessible database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature. UniProt Knowledgebase (UniProtKB) is a protein database partially curated by experts, consisting of two sections: UniProtKB/Swiss-Prot (containing reviewed, manually annotated entries) and UniProtKB/TrEMBL (containing unreviewed, automatically annotated entries). As of 19 March 2014, release "2014_03" of UniProtKB/Swiss-Prot contains 542,782 sequence entries (comprising 193,019,802 amino acids abstracted from 226,896 references) and release "2014_03" of UniProtKB/TrEMBL contains 54,247,468 sequence entries (comprising 17,207,833,179 amino acids).
The UniProt consortium comprises the European Bioinformatics Institute (EBI), the Swiss Institute of Bioinformatics (SIB), and the Protein Information Resource (PIR). EBI, located at the Wellcome Trust Genome Campus in Hinxton, UK, hosts a large resource of bioinformatics databases and services. SIB, located in Geneva, Switzerland, maintains the ExPASy (Expert Protein Analysis System) servers that are a central resource for proteomics tools and databases.
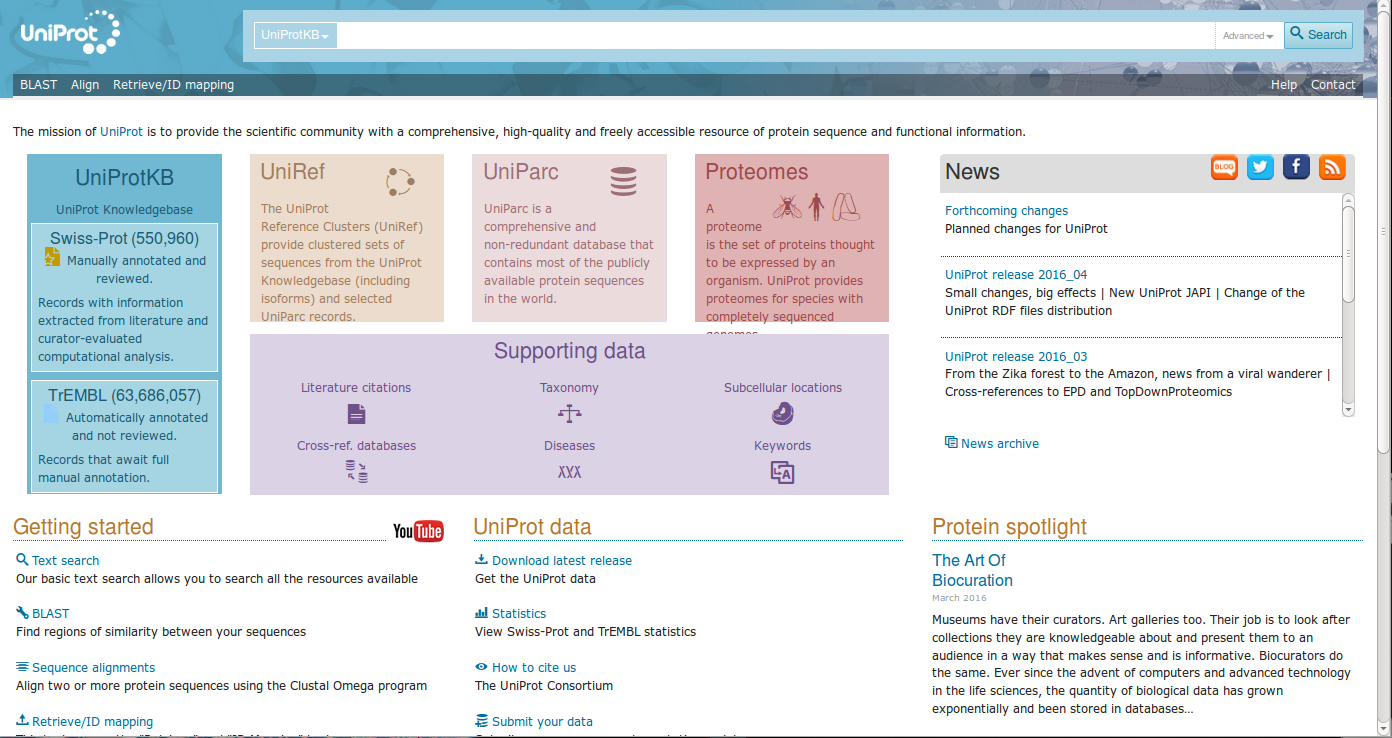
Uniprot collects a large number of different proteins, each entry is associated to an unique "Uniprot identification number".
Now, try to open one "entry", for instance P40337 (Human von Hippel-Lindau protein).
Data are organized by different sections, such as Function, Names & Taxonomy, Subcellular location, Pathology & Biotech, ecc
On the webpages of computers scattered around the world there are many "applications", or web server, that we can use to make Bioinformatics calculations. The web servers are nothing more than dynamic web sites where we fill the appropriate fields with our data, and after waiting for a short time, the Web server will then presents us a new web page with results. The web servers that we will use during our tutorials are numerous and range from the famous Google (to search other pages with information of interest) to web services that can be used to translate, identify, align and analyze nucleotide sequences or protein sequences.
During our tutorials we will use either several web server on line and different stand-alone programs (i.e. installed on the local machine). All these web servers and programs work with linux operating systems, as well as with both Windows and Mac operating systems. In other words, you can try these programs and practise the same exercises from your home computer connected to Internet. Here, a list of the main web servers and programs that we will see during our exercises. Some examples:
To make an example of the usefulness of these online services, try inquiring some web servers looking for information about a well known protein, such as hemoglobin. Later, we will see in more detail the possibilities and how interpret results from different servers:
What is Europe PMC? Europe PMC is a repository, providing access to worldwide life sciences articles, books, patents and clinical guidelines. Europe PMC provides links to relevant records in databases such as Uniprot, European Nucleotide Archive (ENA), Protein Data Bank Europe (PDBE) and BioStudies. ".
The repository offers several tools to perform bibliographic searches. Among them, the most important is the possibility to automatically highlight relevant keywords within several abstracts. Now, try to search breast cancer (a generic definition for different cancer subtypes).

Several papers are sorted by different categories, such as Relevance, Date, Time Cited, while a box with Popular Content Sets is located on the right side.
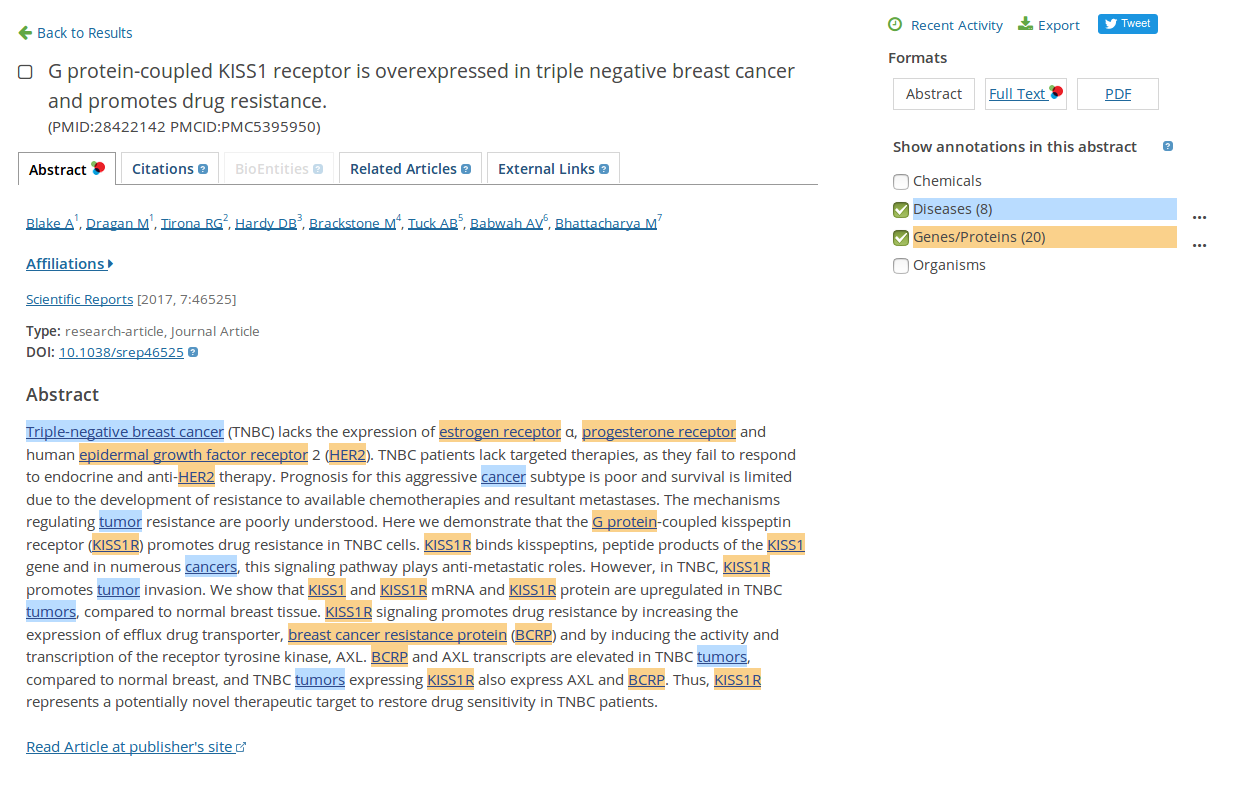
Now, please open the first result (If you have found no relevant information, please try with this example: G protein-coupled KISS1 receptor is overexpressed in triple negative breast cancer and promotes drug resistance).

Using the box on the right side you can highlight different relevant keywords
Dynamic programs, such as Smith and Waterman, are ideal to aligns two sequences accurately, but they are too slow to perform similarity searches in databases. A modern personal computer perform a complete alignment in few minutes, but considering a large database, as GenBank that contains millions of sequences, then the time for execution may require many hours. For example, if you wanted to make a GenBank search for each gene identified in the genome of an entire organism, e.g. yeast with its 6000 genes, you should wait for several years to complete the analysis. Therefore, in order to perform similarity searches using large database faster programs are nedeed. The first program that really performed fast similarity search also using large databases has been FASTA , originally developed by Lipman and Pearson in 1985. This algorithm is based on an indexing words strategy, an intuition that speed up searches
FASTA can be used via web server from EBI website.
The program also introduced a precise format (FASTA format), nowaday commonly used to describe amino acid sequences FASTA (for more information please scroll down this page, see Turquoise Box).
When you have your translated sequence in FASTA format, align it through FASTA, against the UniProtKB/SwissProt database. Analyze your results and try to get the UniProt ID for your unknown protein. To note: Waiting time may vary depending on current server load.
Once obtained the proteins name, through Expasy, you can determine the nature of the protein. To this aim, simply enter in the search form of UniProt the accession number retreived using psi-blast (see next exercise) or from fasta (use the sequence name lying in field: Primary accession number).Paste your amino acid sequence into input box labeled "Search"; as database, please select ("Choose databases" option) "nr" (one of the database of proteins). The other parameters should not be changed.
Click on "BLAST" to send your request.
This creates a temporary wait page, alignment against large databases may takes a long time.
After waiting few minutes (as indicated) it should pop up a new window containing your results.
If the server has not already finished, a waiting message is automatically updated. To Note: the waiting time may vary depending on the server load.
A lot of information can be obtained by clicking on the alignment
and looking at the various fields provided by the program. Spend a little bit of time
to understand if and how is easy to find information on the sequence that you provided as input.
In the page header, at top right (link "Show conserved domains ") you can open a window highlighting the domains found by program for the query sequence. By clicking on the image you can get more information on sequences that present domains. The results page includes, five parts (top to bottom). A entirely equivalent organization is for psi-blast results.
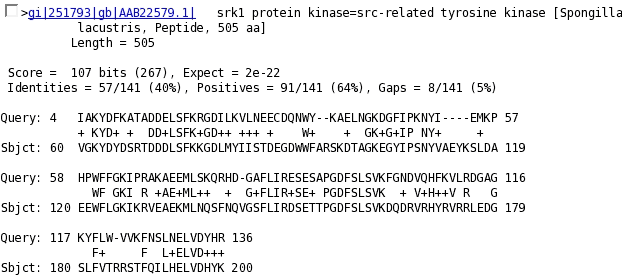
Subsequently the alignments with the sequence "query" are shown. For each alignment is indicated:
Please repeat both the Fasta and Blast searches using the nucleotide sequence of your unkown protein (one for each student).
Please, open NCBI and select blast; Repeat the search using psi-blast and try to find out the differences among the two software.Paste your sequence into input box labeled "Search"; as database, please select ("Choose databases" option) "nr" (one of the database of proteins). The other parameters should not be changed.
Click on "BLAST" to send your request.
This creates a temporary page, please note that align a sequence against a large databases may require long time.
After waiting few minutes (as indicated) it should pop up a new window containing your results.
If the server has not already finished, a waiting message is automatically updated. To Note: the waiting time may vary depending on the server load.
Search for an article in the biomedical field can be done via the PubMed database NCBI. Let's try retrieving the scientific publication on the human genome sequencing:
Now let's use the same keywords but doing more targeted research.
This time the list of retrieved items is shorter. The reference that we were looking for is "Nature. 2004 Oct 21; 431 (7011): 931-45"; Click on it to read the abstract. Selecting the image with the words "nature" you will be redirect to the Publisher's website where you can download the article in electronic format (PDF).
Now, search the electronic version (PDF format, printable) of an article on the bacterium E. coli genome sequencing. The right reference is:
Welch et al. (2002), PNAS, 99 (26), 17020-4
Bibliographic references are generally organized (not necessarily in this order) by author or authors et al., year of publication, journal abbreviation (in this example: Proceedings of the National Academy of Sciences of the United States of America), often in italics. Optionally, volume number followed by the file number (in brackets, often bold).
Since we know the exact bibliographic reference of the article, we do not need to look for him in PubMed. Now, we have to check if the University has the subscription to download this article in electronic format.
* FASTA format: is one of the several way we can use to represent biological sequence. It is simple and easy to read.
The first row is always preceded by '>' followed by several information, e.g sequence name, gene code, official identification number, database;
the second line and over are for the sequence.
An example:
>envelope protein (TVFV2E)
ELRLRYCAPAGFALLKCNDADYDGFKTNCSNVSVVHCTNLMNTTVTTGLLLNGSYSENRT
QIWQKHRTSNDSALILLNKHYNLTVTCKRPGNKTVLPVTIMAGLVFHSQKYNLRLRQAWC
HFPSNWKGAWKEVKEEIVNLPKERYRGTNDPKRIFFQRQWGDPETANLWFNCHGEFFYCK
MDWFLNYLNNLTVDADHNECKNTSGTKSGNKRAPGPCVQRTYVACHIRSVIIWLETISKK
TYAPPREGHLECTSTVTGMTVELNYIPKNRTNVTLSPQIESIWAAELDRYKLVEITPIGF
APTEVRRYTGGHERQKRVPFVXXXXXXXXXXXXXXXXXXXXXXVQSQHLLAGILQQQKLL
AAVEAQQQMLKLTIWGVK