Quick Help and References |
|
| Description | |
|
Espritz is a new web server to detect regions of proteins which
are thought to contain no structural content. This non-structure is known as protein disorder.
Espritz uses an efficient and accurate prediction algorithm based on Bi-directional Recursive Neural
networks (BRNN's). BRNN's are a sequence based machine learning algorithm which have being found to be useful for other structural predictions
[1,2].
The method is based solely on the sequence and does not use expensive computations to find multiple sequence alignments. Espritz also calculates some interesting statistics about all proteins submitted and on each protein individually. For a slightly more detailed technical description of the capabilities and technology used, see the methods page. For the most detailed description see our paper. | |
| Input (Single mode) | |
| E-Mail address | |
|
This is not a requirement. However, if you tick the "E-mail
notification of results" box then make sure your email address is
typed correctly. If no email is supplied do not close the window
(unless bookmarked) while waiting for the results.
| |
| Sequence Name | |
|
An optional title for your submission. This will appear in the header
of the output. We suggest you select one.
| |
| Sequence | |
All sequences submitted must be in fasta format.
Invalid amino acid types, spaces, return characters should be ignored by the server. However, we suggest that you keep the input in the
following format:
>P1 MIETPYYLIDKAKLTRNMERIAHVREKSGAKALLALKCFATWSVFDLMRD YMDGTTSSSLFEVRLGRERFGKETHAYSVAYGDNEIDEVVSHADKIIFNS ISQLERFADKAAGIARGLRLNPQVSSSSFDL >P2 MTFSKELREASRPIIDDIYNDGFIQDLLAGKLSNQAVRQYLRADASYLK EFTNIYAMLIPKMSSMEDVKFLVEQIEFMLEGEVEAHEVLADFINE >P3 MENKKMNLLLFSGDYDKALASLIIANAAREMEIEVTIFCAFWGLLLLRD PEKASQEDKSLYEQAFSSLTPREAEELPLSKMNLGGIGKKMLLEMMKEE KAPKLSDLLSGARKK >P4 MKKVFYPLACCCLAAGVFASCGGQKKANAQEEPSKVALSYSKSLKAPET DSLNLPVDENGYITIFDwhere P1, P2, P3 and P4 are the sequence identifiers. It is vital that all identifiers are unique. For an example multiple fasta example see here and for an example single fasta see here. Once your sequence(s) are in fasta format there are two input types:
| |
| Options | |
ESpritz The predictor is trained on three types of disorder predictions:
X-ray, disprot and NMR disorder types can be chosen using the left most drop down menu in the option menu. ESpritz produces a probability of disorder for each residue. A decision on what probability cut-off threshold produces the best disorder was made on the training sets for each predictor. Although, our method produces true Bayesian probabilities the data used to learn the disordered regions was imbalanced and therefore using 0.5 as the probability decision is not recommended. For this reason internal probability thresholds are defined on each predictors training set. The server hides this information from the user by allowing two types of threshold with respect to the training dataset:
| |
| Output (Global stats) | |
| The first page the user sees is a global statistics and download page. | |
| Statistics on all proteins | |
The global statistics are in the following form: Total amino acids : ℕ Total proteins : ℕ Total % disorder : ℝ % of proteins with at least 1 disordered region greater than 30 amino acids : ℝ % of proteins with at least 1 disordered region greater than 50 amino acids : ℝ Total no. of disordered regions greater than 30 amino acids : ℕ Total no. of disordered regions greater than 50 amino acids : ℕ Number of disordered segments : ℕ Mean segment length (standard deviation) : ℝ (ℝ) | |
Residues are split into 3 groups charged, uncharged and hydrophobic. The occurences and disorder percentages are reported for the 3 groups. 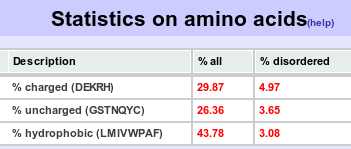 In the example above 29.87% of the residues in all the proteins submitted are charged. Also, of this 29.87% residues 4.97% are disordered. In addition, for completeness this table is expaned to include the percentage occurence of all the 20 amino acids and its percentage disorder. |
|
| Available files for download | |
Available for download is a Disorder.tar.gz archive containing:
| |
|
Distribution of disordered segments: The final graph to download is the Histogram of disordered segments graph. An example graph
is provided below. The x-axis contains the disorder segment ranked by its length.
The y-axis contains the length of the disorder segment. | |
Distribution amino acids: Also available to download is the distribution of amino acids in any state and in a disordered state.
An example graph is provided below. The x-axis contains the amino acid under investigation.
The red bars show the percentage that amino acid occurs. The blue bar shows the percentage of the red bar is disordered.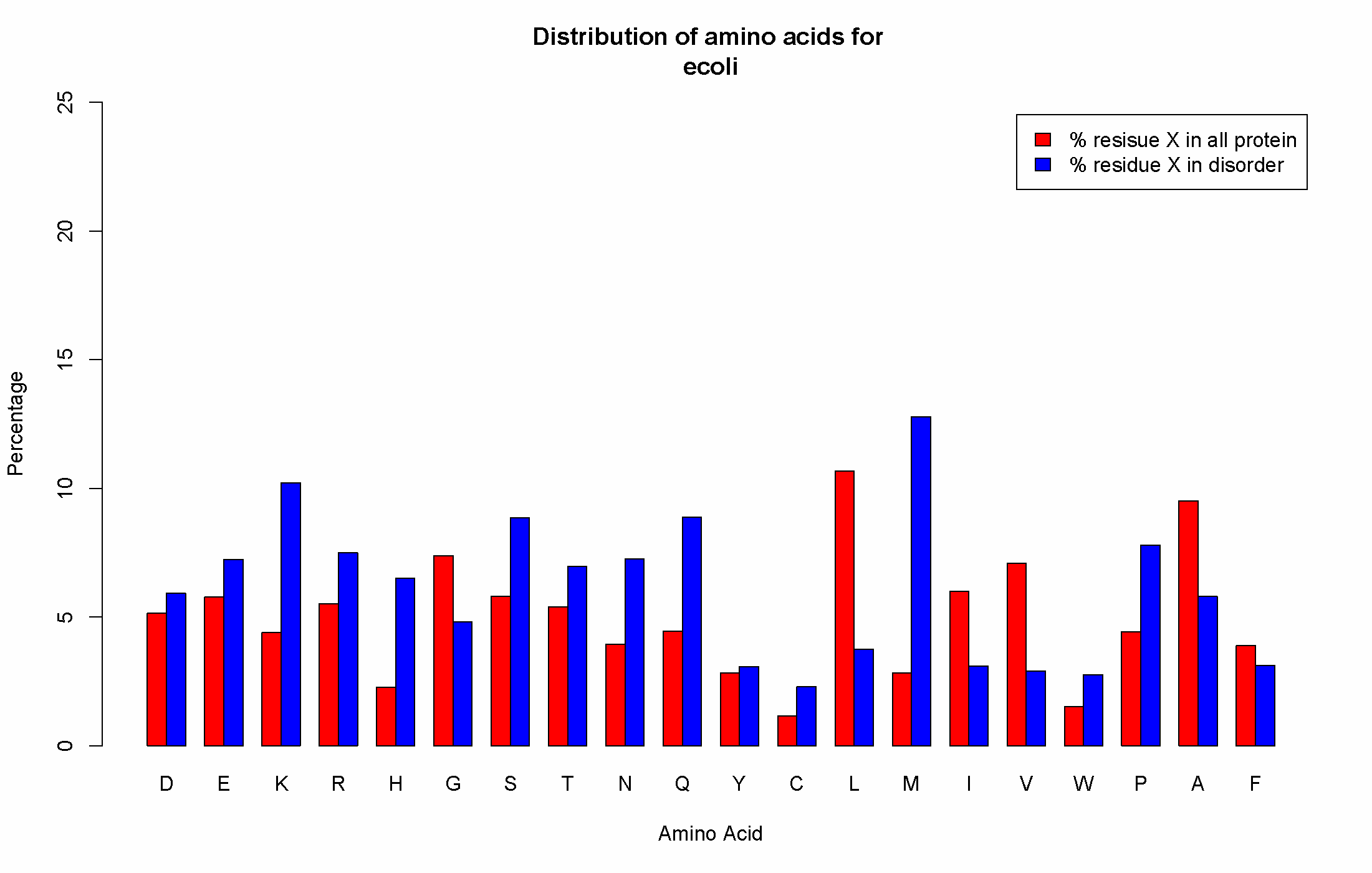
| |
| Links to individual protein pages | |
Finally, links to the individual proteins are available (see next section). An example list is shown below. 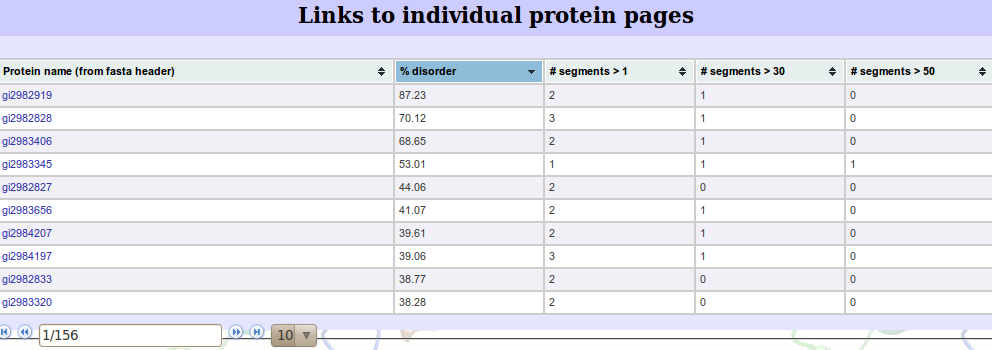 We feel it is important to rank this list considering it may be a very large list. We allow rankings by %disorder, number of segments, number of segments greather than 30 and number of segments gretaer than 50. Each page of the list contains 10 proteins where the next 10 can be accessed by clicking the next arrow button. | |
| Output (individual protein) | |
|
The output page presented for each protein gives a quick overview of the proteins disorder and statistics. In addition,
secondary structure and linear motifs are presented for each disorder segment.
|
|
| Available files and links | |
Total amino acids: Total % disorder: Total no. of disordered regions greater than 30 amino acids: Total no. of disordered regions greater than 50 amino acids: Number of disordered segments: Length distribution of segments (N to C terminal order): |
|
| Disordered residues and stats: | |
This panel consists of two pieces of information:
|
|
| Examples | |
|
Here we present 1 single fasta pasted example, all the casp 9 targets pasted and the human genome downloaded from ftp://ftp.ncbi.nlm.nih.gov/genbank/. This section should also serve as a tutorial of how to use ESpritz. Example 1 - Human p53. UniProt ID: P04637, 393 residues calculations with short option. Input: input page for human p53. Multiple protein example 2 - Entire casp9 disorder targets. 117 proteins. Download the casp9 fasta sequences, hit browse on the input page and upload the file. This example shows how to process multiple sequences.
Genome example 4 - Entire
proteome of homo sapien. 39151 proteins. Download the human fasta sequences, hit browse on the input page and upload the file. This took approx. 6.5 hrs on
the current infrastructure.
| |
| References | |
Please cite:
I. Walsh, A. J. M. Martin, T. Di domenico, S. C. E. Tosatto | |