Esercitazione - Parte 3
Struttura terziaria
In questa esercitazione vedremo che tipo di informazioni possiamo ricavare partendo dalla struttura terziaria. La struttura terziaria, conosciuta anche come struttura tridimensionale (o 3D) viene risolta attraverso metodiche sperimentali quali la cristallografia e la NMR. Come di certo saprete, con l'analisi della struttura terziaria ci è possibile studiare l'organizzazione degli elementi strutturali di una proteina nei tre piani dello spazio. Ovviamente con una struttura proteica possiamo fare molto di più... di seguito alcuni esempi.
Visualizzazione della struttura proteica
Scopo di questa parte è visualizzare in 3D la struttura di una proteina e seguente analisi del sito attivo.
Per visualizzare il file pdb si può utilizzare online il programma JMol oppure nel nostro caso il programma PyMol già installato nel computer (cercatelo nel menu con i programmi).
I codici PDB delle proteine da analizzare sono:
Proteina 1: 1yso
Proteina 2: 1dsw
In entrambi i casi la proteina è una superossido dismutasi (SOD), un enzima con un atomo di rame e uno di zinco nel sito attivo.
Per prima cosa dovete scaricare i file pdb dalla banca dati di strutture cristallografiche RCSB PDB sul proprio computer. Aprite la pagina di RCSB e inserite il codice del file pdb nel campo a destra di "PDB ID or Keyword" e premete Search. Comparirà la pagina relativa alla proteina data con a destra anche l'immagine della proteina. Selezionate "Download files/PDB file (text)" e salvate il file sul vostro computer.
Uso di PyMol:
- aprite il file PDB con la struttura della proteina salvata con File/Open
- tenendo premuto il tasto sinistro del mouse e muovendolo si può ruotare la proteina.
- con il tasto destro è possibile zoommarla, mentre con il tasto centrale la si puà spostare su/giù/sinistra/destra. La rotellina del mouse modifica i piani anteriore e posteriore che "tagliano" la molecola parallelamente allo schermo.
- nella parte destra del programma c'è l'elenco degli oggetti visualizzati (molecole o selezioni/parti di molecole) ed i pulsanti a tendina A, S, H, L ed C che stanno rispettivamente per Azione, Show (mostra), Hide (nascondi), Label (etichetta) e Colore. Proviamo ad usarli:
- nascondiamo tutta la proteina: sotto all, premiamo H e poi everything
- mostriamo la proteina in maniera semplificata (eliche e freccie per foglietti beta): sotto il codice PDB della proteina, premiamo S e poi cartoon. Potete provare a nascondere (H) e mostrare (S) anche diverse rappresentazioni della proteina tipo sticks (mostra soltanto i legami tra gli atomi) o spheres (con sfere per atomo)
- coloriamo la proteina per tipo di struttura secondaria: proteina/C/by ss/scegliete i colori
Visualizzazione dei metalli nel sito attivo:
- ora selezioniamo gli atomi di Cu e Zn e mostriamoli come sfere. Per prima cosa mostriamo la sequenza intera della proteina con Display (nel menù principale di PyMol)/Sequence. Comparirà la sequenza che è cliccabile e selezionabile. Scorrete la sequenza ed alla fine, prima delle molecole d'acqua (indicate con O, per ossigeno - gli H sono in genere invisibili ai raggi X) ci sono i due metalli e selezionateli. Comparirà un nuovo oggetto chiamato (sele) per selezione. Rinominate (sele) con A/rename selection e dategli il nome "cofattori"
- ora disegnate i cofattori come sfere (S/spheres) e colorate per tipo di atomo (C/by element)
- aggiungiamo anche il nome dei due cofattori con L/atom name. Poiché l'etichetta verrà scritta nel centro dell'atomo, ovvero all'interno della sfera che lo rappresenta, l'etichetta non è visibile. Possiamo sposare le etichette un po di lato digitando set label_position,(2,2,2) nella riga di commandi di PyMol (dove lampeggia il cursore, dopo PyMOL>). A questo punto i nomi degli atomi dovrebbero essere visibili.
- potete zoomare sui cofattori (o sulla proteina intera) scegliendo A/zoom
Ora calcoliamo la distanza tra i due atomi: nel menù scegliete Wizard/Measurement/selezionate il primo atomo e poi il secondo ed infine premete Done. Comparirà un nuovo oggetto chiamato measure01 con la distanza tra i due atomi in Angstrom come etichetta.
Riportate la distanza tra gli atomi che appare nel disegno sul foglio con le domande (ma non chiudete PyMol che tra poco vi servirà di nuovo!).
Adesso cercheremo i ligandi dei due atomi (ovvero quali amminoacidi della proteina legano i metalli del sito attivo) utilizzando il server PDBsum. Inserite il codice PDB della vostra proteina e premete Find.
Nel sito potete vedere diverse informazioni, come ad esempio la reazione enzimatica, la struttura secondaria (sotto il link Protein chain) e i ligandi dei cofattori (sotto Metal ions).
Cliccando sul link dei cofattori appare il disegno degli amminoacidi che legano gli ioni metallici (ignorate la molecola d'acqua); segnatevi i corrispettivi nomi (es His46 ...). Tenete in conto sia i legami diretti che i ponti H. Per interpretare il disegno di LIGPLOT basatevi su questa leggenda e descrizione:
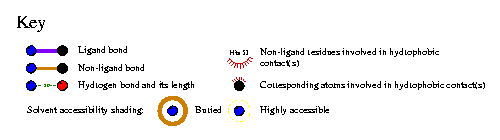
- Ligand bonds are shown with thick lines (here in purple).
- Non-ligand bonds (ie belonging to those protein residues to which the ligand is hydrogen-bonded) are shown with thin (orange) bonds.
- Hydrogen bonds are shown by dashed (green) lines with the length of the bond printed in the middle.
- Hydrophobic contacts between protein and ligand are indicated by the (brick-red) spoked arcs. Protein residues involved only in these contacts with the ligand are shown by a single spoked arc. Other atoms involved in hydrophobic contacts have spokes coming out of the individual atoms.
- Atomic accessibilities are (optionally) shown by shaded circles around the atoms. The thicker and darker the circle, the more buried and solvent inaccessible the atom. Exposed, highly accessible surface atoms are surrounded by a thinner, lighter circle.
- Chain identifiers (where relevant) are shown in parentheses after the residue names.
Ora visualizziamoli anche in PyMol, per fare questo selezionate i residui interessati dalla cliccando sulla sequenza, rinominate la selezione (A/rename) e mostrate questi residui come legami (S/sticks) colorati per tipo di atomo (C/by element). Notate che gli atomi che legano i metalli sono soprattutto gli azoti "epsilon" dell'anello imidazolico delle istidine.
Se avete seguito bene le istruzioni, il risultato finale dovrebbe essere una rappresentazione di questo tipo:
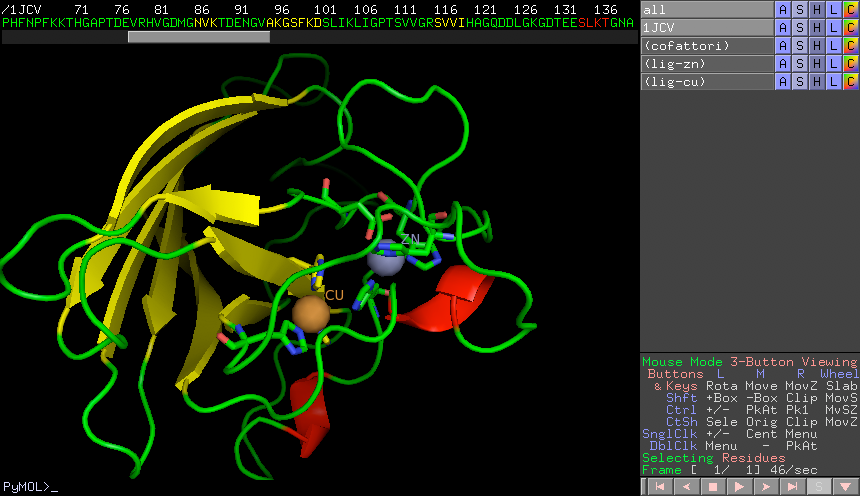
Il programma PyMol è molto potente e premette di realizzare immagini di livello elevato, addatto alle pubblicazioni. Tuttavia per utilizzarlo al meglio bisogna conoscere i suoi numerosi commandi. Un'esempio di illustrazioni fatte con PyMol e di istruzioni su come farle le potete trovare dal sito PyMol tutorials.
Le proteine visualizzate in questa esercitazione sono volutamente semplici per poter identificare facilmente quello che ci interessa.
Se volete visualizzare strutture più complesse ecco una lista di proteine "consigliate". Queste e tutte le altre proteine la cui struttura 3D è stata determinata, sono depositate nella banca dati RCSB Protein Data Bank. Inserite nel campo di ricerca il codice PDB della proteina. Dopo aver effettuato la ricerca potete visualizzare la molecola in JMol direttamente cliccando sul link sotto l'immagine a destra oppure scaricare il file PDB sul disco tramite Download files/PDB file dal menù di sinistra. I file PDB scaricati sul disco potete visualizzarli anche da casa con computer con Windows utilizzando il programma gratuito RasMol (RasMol Latest Windows Installer)
oppure PyMol visto oggi.
- Nucleosoma (1aoi), 2 giri di DNA intorno a proteine istoniche (ottamero)
- Ribosoma subunità maggiore 50S, (1ffk), proteine in CA, RNA con tutti gli atomi
- Ribosoma subunità minore 30S, (1fka), alcune proteine in CA, RNA con tutti gli atomi
- Fotosistema I (1jb0), proteina con tantissimi cofattori (clorofille, carotenoidi ecc.)
- TATA binding protein legata alla regione "TATA box" del DNA, da notare la piega del DNA (1ytb)
- Emoglobina Deoxy (2hhb)
- Emoglobina Oxy (1hho), unità assimetrica ha solo due catene, scaricate l'unità biologica che ha 4 catene
- Batteriofago phiX174 (1cd3), un 120 del capside. Per scaricare il capside intero, scaricate l'unità biologica - Attenzione: si tratta di 10 MB di file compressi (che diventano 40 MB)
- Porina (2por), scaricare l'unità biologica per il trimero intero
- Chaperonina HSP-60, GroEL/GroES (1aon)
- Anticorpo IGG (1igt)
- B-DNA (1bna), destrogira, la forma di Watson e Crick
- A-DNA (1ana), destrogira, basi inclinate, in condizioni deidratanti, forma tipica dell'RNA
- Z-DNA (2dcg), levogira, in condizioni di alta salinità e con coppie GC alternate
- tRNA con fenilalanina (4tna)
- tRNA con aspartato (2tra)
Sempre nel sito RCSB PDB, si trova una sezione didattica e molto interessante chiamata "Molecule of the month" dove ogni mese viene descritta la struttura e funzione di una proteina di interresse biologico, corredata da belle immagini e istruzioni su come visualizzarle utilizzando i file PDB scaricabili dal sito e RasMol oppure JMol (online).
Predizione della struttura
Un programma sviluppato dall'università di Padova in grado di predirre la struttura proteica dalla sua sequenza amminoacidica è Homer; questo server cerca tramite diverse iterazioni di PSI-BLAST il modello/templato migliore e su questo modello 3D calcola la struttura terziaria della sequenza data, producendo una struttura visualizzabile tramite Jmol.
Poiché i calcoli richiesti per ottenere il risultato sono parecchio pesanti dal punto di vista computazionale, vi forniamo direttamente le informazioni per aprire i risultati precalcolati; riuscite a risalire a quale proteina appartiene questo modello?
Andate alla seguente pagina per visualizzare il risultato.
Col tasto "Template" viene visualizzata la struttura della proteina di struttura nota, usata per predirre il ripiegamento della sequenza query visibile con il tasto "Model".
"Model quality" visualizza e colora il modello 3D della vostra query secondo l'affidabilità della predizione (blu -> maggiore affidabilità, rosso -> minore affidabilità).
Il collegamento PDF (grafico) o TEXT (elenco numerico) di "FRST energy profile of the modelled structure", mostra l'affidabilità della predizione lungo la catena amminoacidica. I valori sotto lo 0 indicano regioni predette in maniera affidabile, viceversa per quelle maggiori di 0.
Allineamento strutturale
Come le sequenze aminoacidiche (struttura primaria) si possono allineare tra di loro, in modo analogo si possono allineare anche le strutture terziarie ovvero le strutture tridimensionali di proteine diverse, massimizzando il grado di sovrapposizione tra gli atomi delle due proteine (quantificato dal valore di RMSD). Uno dei programm più usati è CE che sta per il nome dell'algoritmo usato: Combinatorial Extension.
In questo esercizio proveremo ad allineare strutturalmente la proteina glutatione perossidasi (gpx) 1 con due suoi omologhi (anche strutturali): gpx 4 e gpx 5.
Codice PDB della proteina gpx 1 è 2f8a
Struttura 1: 2gs3 (gpx 4)
Struttura 2: 2p5q (gpx 5)
Per prima cosa scaricate dal sito RCSB PDB i due file PDB da allineare. Aprite la pagina di CE, andate su "Two chains", usate l'opzione "User file" e tramite il pulsante "Browse" spedite al server i due file di struttura da allineare (prima la gpx 1 e poi la gpx 4 o 5) e premete "Calculate alignment". Una volta calcolato l'allineamento, il server vi risponde con l'allineamento delle sequenze (da allineamento strutturale!!!), il valore di RMSD (indice della discrepanza media tra le posizioni degli atomi allineati) e da l'opzione di scaricare il file PDB con le due strutture sovrapposte sotto "Download alignment as a PDB file". Cliccate su tale link e scaricate il file PDB sul disco rinominandolo in ce.pdb (destro click sul file/rinomina).
Ora cerchiamo di visualizzare le due strutture sovrapposte con PyMol. Aprite PyMol e caricate il file ce.pdb. Poiché tale file PDB contiene due modelli, PyMol ne mostra soltanto il primo. Per mostrarli entrambi, digitate il comando "split_states ce" senza virgolette al prompt di PyMol (dopo PyMOL>). PyMol mostrerà questa volta le due catene separate (chiamatte ce_0001 e ce_0002). Ora cancellate la prima molecola (chiamata ce) con ce/A/delete object, nascondete tutto con all/H/everything e mostrate le due catene come linee che collegano soltanto i carboni alfa (tralasciando le catene laterali) con ce_0001/S/ribbon e colorate la catena di rosso con C/reds/red. Mostrate e colorate di verde l'altra catena. Mostrate le sequenze delle due strutture con Display/Sequence.
Come potete vedere, le due proteine hanno una struttura molto simile anche se hanno una bassa similarità di sequenza. Cliccando sulla catena verde (ovvero la struttura della gpx 4/5 allineata sulla rossa gpx 1) ed aiutandovi con la barra delle sequenze, siete in grado di individuare i due loop dove le gpx 4/5 differiscono dalla 1?
Ricerca di funzioni (tramite identificazione di domini)
Provate (se possibile) ad identificare la o le funzioni delle due sequenze di esempio. Se riuscite, provate ad identificare la posizione (numeri degli amminoacidi) in cui si trovano i domini riscontrati. In base a quanto visto nella esercitazione precedente, scegliete il servizio web che più vi sembra adatto per un'operazione di questo tipo.
- Siete riusciti ad identificare questa proteina dal risultato dell'allineamento contro una banca dati?
Localizzazione dei residui conservati
Esaminando una struttura proteica nota (ovvero di cui si conosce la struttura ad alta risoluzione ottenuta tramite tecniche di diffrazione di raggi X oppure NMR) ci si potrebbe chiedere quali sono e dove si trovano gli aminoacidi conservati e quali sono invece quelli con meno vincoli funzionali e che per questo motivo possono evolvere liberamente. Il server ConSurf risponde a queste domande. Questo server riesce, tramite una ricerca nelle banche dati con PSI-BLAST e sucessivo allineamento multiplo di sequenze con MUSCLE oppure CLUSTALW, a stimare il grado di conservazione per ogni aminoacido di una data struttura. Il grado di conservazione degli aminoacidi nella proteina viene visualizzato direttamente sulla struttura query tramite Jmol. Il colore di ciascun residuo varia da celeste (posizioni altamente variabili) a rosso scuro (posizioni conservate in strutture diverse). Le posizioni per le quali non si riesce ad ottenere una stima ragionevole della variabilità sono indicate in giallo chiaro.
Ora provate ad analizzare tramite ConSurf la struttura della deossi-emoglobina. Riempite i campi PDB ID, Chain Identifier e la vostra email per il ritiro dei risultati. Una volta premuto il tasto Submit verrà visualizzata la pagina temporanea che si aggiorna ogni 30 secondi. Entro una decina di minuti dovrebbe comparire la pagina dei risultati finali con il collegamento a Jmol (View ConSurf Results with FirstGlance in Jmol).
Osservate la posizione degli aminoacidi conservati.
Riuscite a dare una spiegazione sul motivo per cui sono proprio quelli gli aminoacidi conservati?
Lo stesso risultato, anche se meno evidente, si ottiene analizzando le altre due catene dell'emoglobina (beta): B oppure D. Poiché le 2 catene alfa (chiamate A e C) nel file PDB sono identiche, potete attivare la visualizzazione anche dell'altra catena attivando i campi sotto "Act on identical chains".
Sequenza: Deossi-emoglobina, codice PDB: 2hhb, catena A oppure C (catene alfa)
Analisi delle proprietà elettrostatiche e predizione degli effetti di mutazioni
BLUUES è un server che ci permette di calcolare il potenziale elettrostatico di una proteina. Tra le varie funzioni di questo utile tool è prevista anche la possibilità di studiare gli effetti di mutazioni. Il programma sfrutta l'equazione di Poisson-Boltzmann di cui ne calcola un'approssimazione lineare, permettendo un calcolo veloce senza perdere precisione.
Vediamo ora come utilizzarlo. Proviamo per esempio a calcolare gli effetti di una mutazione sulla proteina pVHL (Von Hippel–Lindau tumor suppressor).
Il programma si presenta come una pagina web nella quale bisogna inserire delle informazioni. Nella pagina di presentazione è possibile calcolare il
potenziale elettrostatico semplicemente inserendo il codice PDB della vostra proteina di interesse. In alto a destra è presente il tab Mutate, selezionatelo per attivare l'interfaccia che vi permette di studiare gli effetti di mutazioni.
Dovrebbe apparirvi un pannello nel quale inserire il codice PDB. Provate ad inserire il codice 1LM8 e selezionate la catena V. Dopo il tempo necessario in cui il programma caricherà l'elenco degli amminoacidi presenti nella struttura cristallografica dovrebbe apparirvi un secondo pannello nel quale indicare la mutazione desiderata.
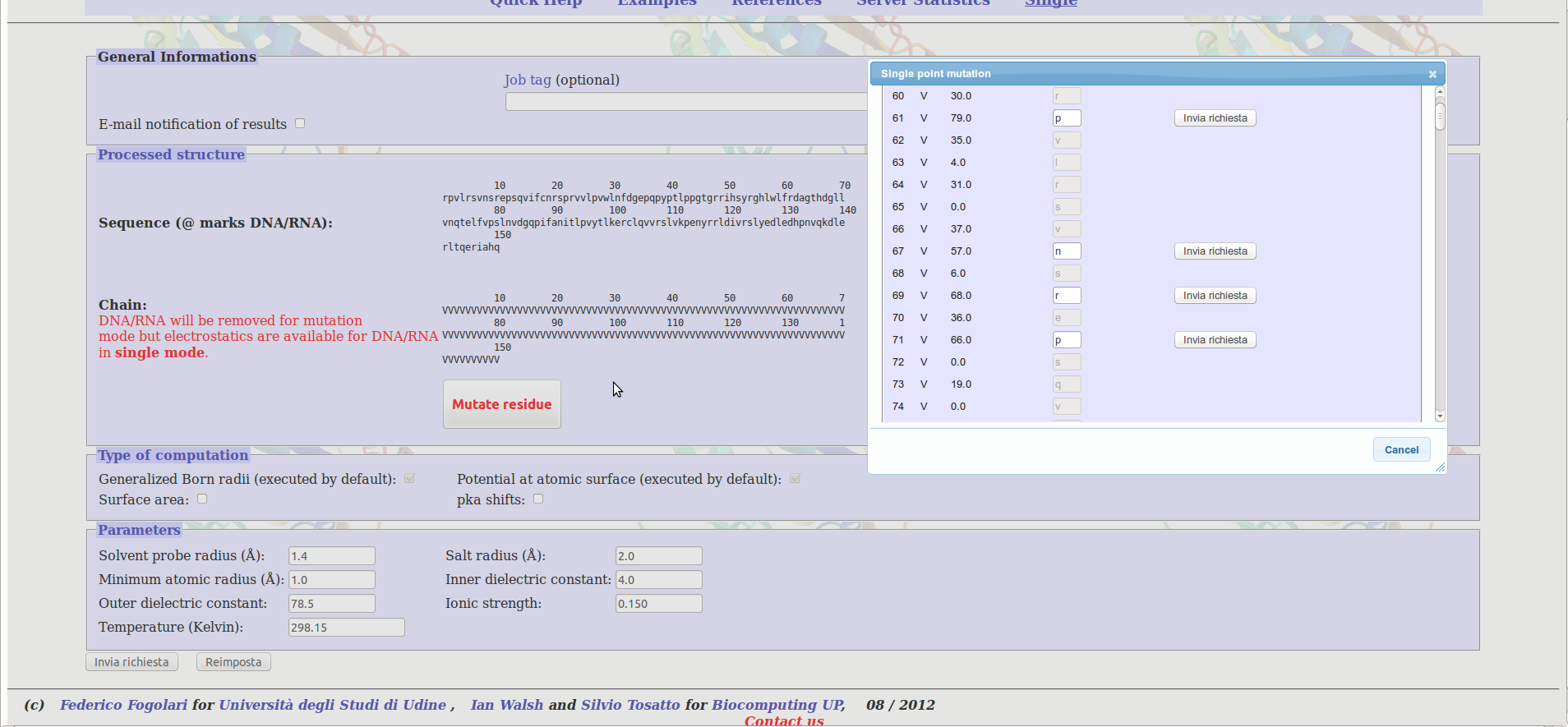
Provate a mutatare uno o più residui a vostro piacimento.
Previsione della struttura secondaria e del disordine
Ci sono dei casi in cui la struttura terziaria non può essere risolta. Pensiamo al caso di proteine contenenti regioni disordinate. In questi casi il programma CSpritz permette di predirre la struttura secondaria della sequenza e fornisce un'indicazione sulle regioni ordinate (strutturate) e su quelle disordinate (non strutturate) della proteina.
Esamineremo due sequenze di criptocromo per verificare i risultati ottenuti tramite gli allineamenti nelle scorse esercitazioni. I risultati vengono spediti via posta entro cinque minuti circa (usate una qualsiasi delle vostre mail); riempite tutti e tre i campi (leggete bene cosa inserire in ogni campo) e scegliete "Long disorder" come tipo di predizione.
La spiegazione del formato di output del risultato lo potete trovare cliccando sulla pagina di aiuto di CSpritz. Osservate bene sia la seconda riga che porta informazioni sulla struttura secondaria che sulla terza che porta informazioni circa l'ordine/disordine.
Interazioni proteina-proteina
Spesso è utile sapere, durante lo studio di una proteina, se essa interagisce con altre e con quale tipo di interazione. Uno dei server che ci può servire in questo caso è STRING. Questo web server ospita la banca dati di interazioni proteina-proteina teoriche (stimate) e quelle conosciute (sperimentali). All'utente viene visualizzata una mappa colorata (grafo, rete di interazione) con la proteina query in centro e circondata da proteine con le quali interagisce. Scegliendo l'opzione "interactive" si possono spostare manualmente i nodi della rete (per facilitarne la visione), oppure tramite i tasti "+ more/- less" si possono inserire o nascondere ulteriori interazioni più lontante.
Sempre partendo da UniProt, visualizzate le reti di interazione tramite STRING delle seguenti proteine:
Sequenza 1: Subunità alfa dell'emoglobina umana
Sequenza 2: Subunità beta dell'emoglobina umana
Che differenze notate?
Provate con altre proteine a vostro piacimento. Che tipi di interazioni avete trovato?
Motivi lineari
I motivi lineari delle proteine sono frammenti funzionali di pochi amminoacidi, che non assumono una struttura tridimensionale definita. Essi indicano siti di modificazioni post traduzionali, siti di legame con vari ligandi o siti di targeting cellulare. Il server ELM cerca di identificarli sulla proteina in esame e ne descrive la loro funzione. Anche se gli ELM (motivi lineari eucariotici) si possono trovare lungo tutta la proteina, essi sono funzionali soltanto nelle regioni non globulari, ovvero disordinate.
Utilizzando il server ELM, provate a identificare i motivi lineari che si trovano nella coda C-terminale disordinata nelle sequenze di criptocromo di piante.
Proteina 1: Q309E8_NICSY (criptocromo di Nicotiana)
Proteina 2: CRY1_ARATH (criptocromo di Arabidopsis)
28-05-2015 10:34:48