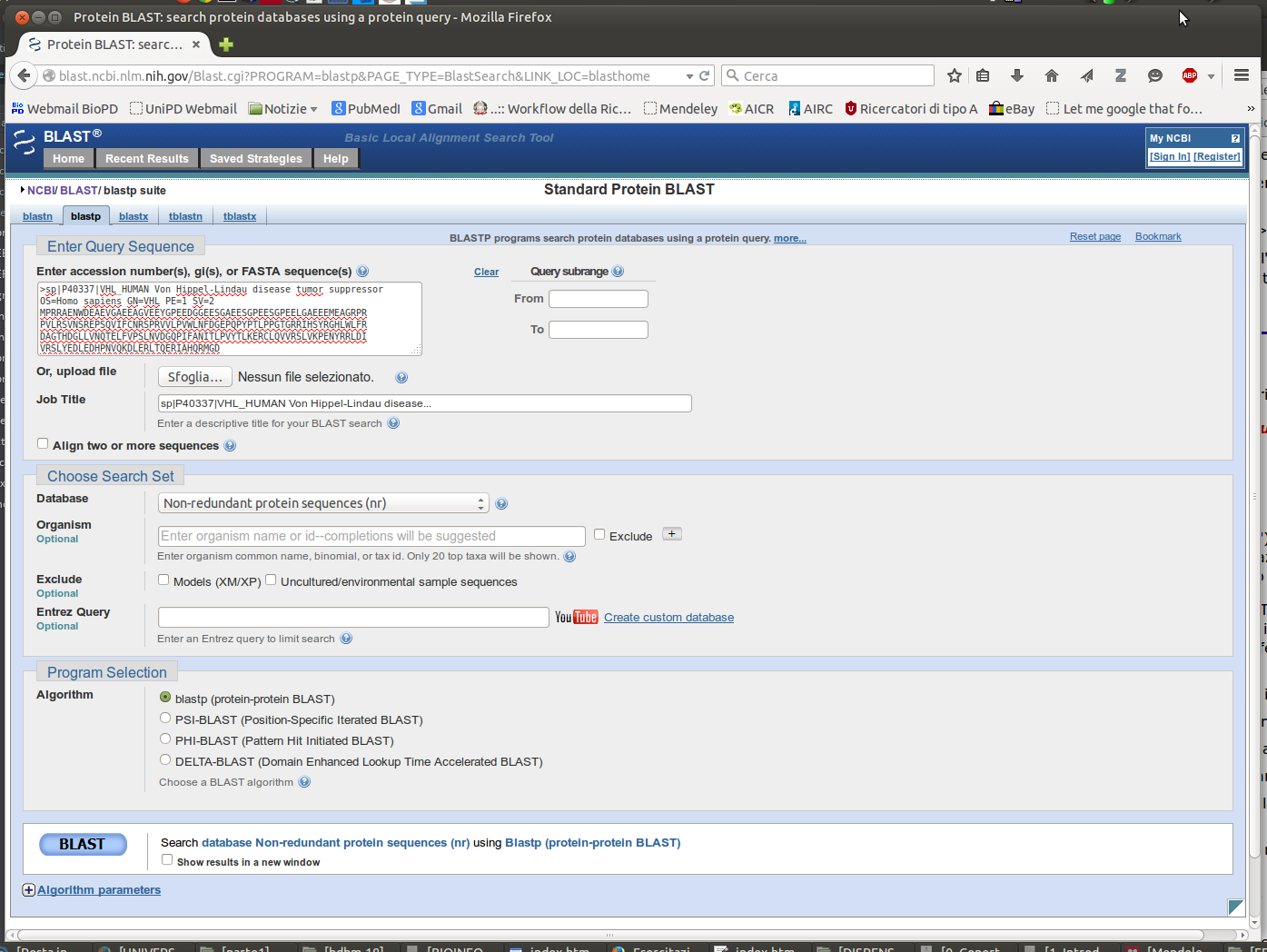
Indentifichiamo la nostra proteina con blastp
Nell'intestazione, in alto a destra (collegamento "Show conserved domains") si può aprire la finestra con che evidenzia i domini trovati dal programma sulla sequenza query. Cliccandoci sopra l'immagine si ottengono ulteriori informazioni su sequenze che rappresentano domini simili. La pagina dei risultati comprende, a parte l'intestazione, cinque parti (dall'alto in basso), del tutto equivalenti a quelle dei risultati di psi-blast.
Dopo pochi minuti dovrebbe apparirvi una schermata simile a questa

- La prima parte dà informazioni sul programma (in questo caso BLASTP), sulla sequenza "query" (quella con cui si effettua la ricerca), tra le quali la sua lunghezza (indicata in aminoacidi), e sul database utilizzati. Presenta inoltre un link denominato "Taxonomy reports", da cui si arriva ad una pagina che presenta tre diverse organizzazioni dei risultati di una data ricerca effettuata con BLAST, in base all'informazione presente nel database tassonomico dell'NCBI.
- La seconda parte consiste in una immagine che illustra graficamente i risultati:
- la linea rossa spessa e graduata rappresenta la sequenza "query";
- i numeri sotto di essa si riferiscono alla sua lunghezza in aminoacidi;
- ciascuna linea sottile sottostante, di vari colori, indica un allineamento della suddetta sequenza con una sequenza del database di proteine;
- il colore di tali linee indica la bontà dell'allineamento, in base alla scala colorimetrica posta all'inizio dell'immagine (rosso - migliore, nero - peggiore);
- passando su ciascuna linea sottile con la freccia del mouse, sul riquadro subito sopra l'immagine compare la descrizione della proteina del database con cui la sequenza query si allinea in quel caso;
- cliccando su tali linee si va invece direttamente all'allineamento (situato nella quarta parte della pagina) tra la "query" e una data sequenza del database.
- La terza parte consiste nell'elenco delle proteine del database scelto che producono allineamenti significativi con la sequenza "query" e comincia con la frase: "Sequences producing significant alignments:". Le sequenze sono ordinate in base all'E value (colonna a destra). Ciascuna sequenza contiene un link (parte sottolineata e in colore), da cui si arriva al record di Entrez relativo a quella sequenza. A destra, per una data sequenza sono inoltre eventualmente presenti i link a Gene ("G").
- La quarta parte visualizza gli allineamenti
significativi della sequenza "query" con sequenze del database scelto e
comincia con la frase: "Alignments".
Data una sequenza del database scelto che produce uno o più allineamenti significativi con la sequenza "query", come prima cosa viene visualizzata la descrizione della sequenza del database, che contiene un link (parte sottolineata e in colore), da cui si arriva al record di Entrez relativo a quella sequenza. La descrizione contiene eventualmente anche il link a Gene ("G"). Sotto la descrizione è indicata la lunghezza in aminoacidi della sequenza.
Proviamo a cliccare nel box dove vengono mostrati i domini conservati individuati
- dovrebbe apparirvi qualcosa di simile a questo:
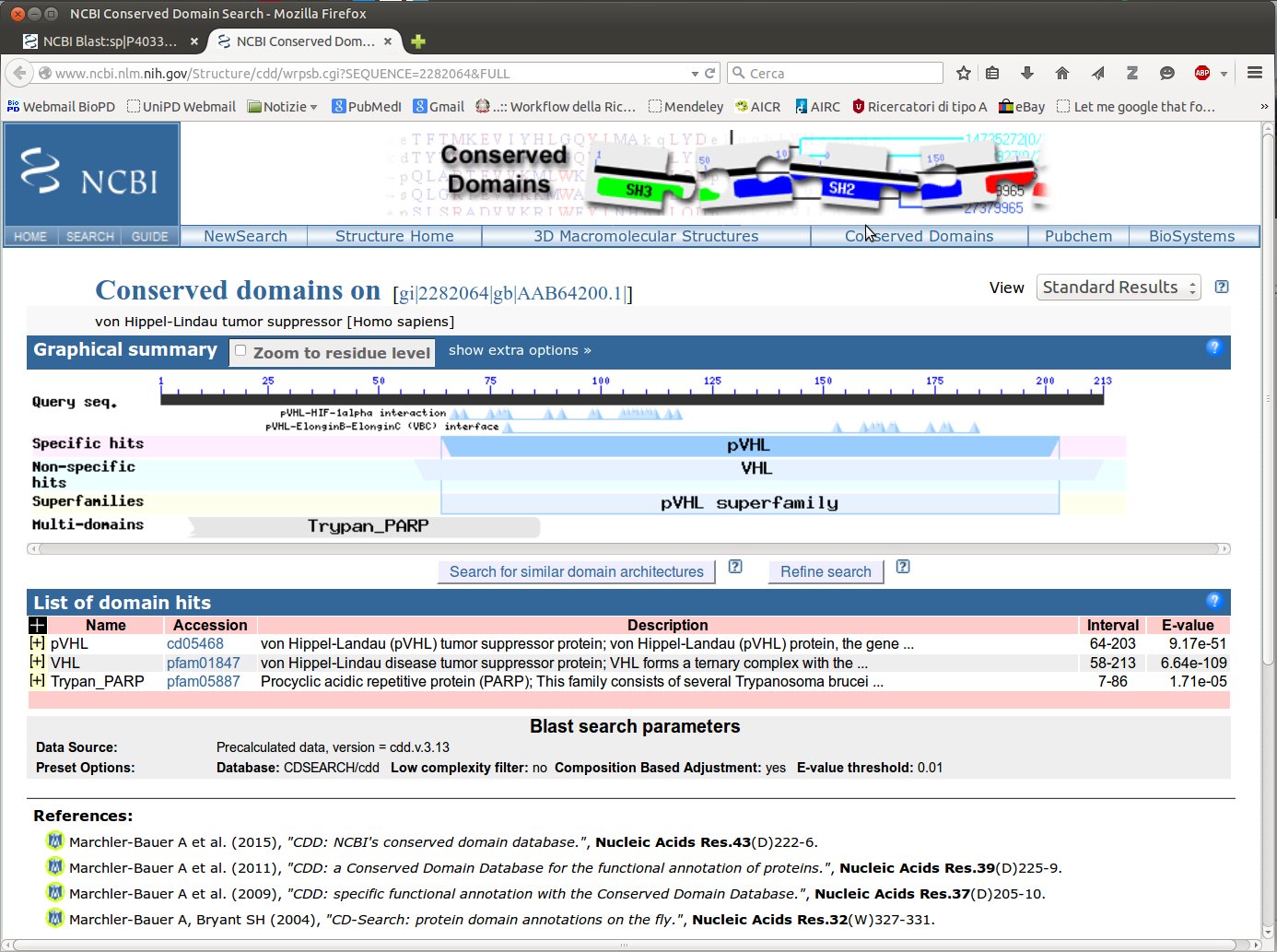
- NOTA: provate a ripetere la ricerca modificando i parametri del programma. In particolare, nella sezione Database selezionate PDB. In questo caso la ricerca verrà effettuata utilizzando il database di strutture tridimensionali e, se presenti, verranno restituite in aoutput le strutture cristallografiche più simili alla vostra proteina.
Analizziamo ora il grado di conservazione della nostra sequenza
Per verificare il grado di conservazione della nostra proteina ci serviremo di un database per la ricerca di ortologhi
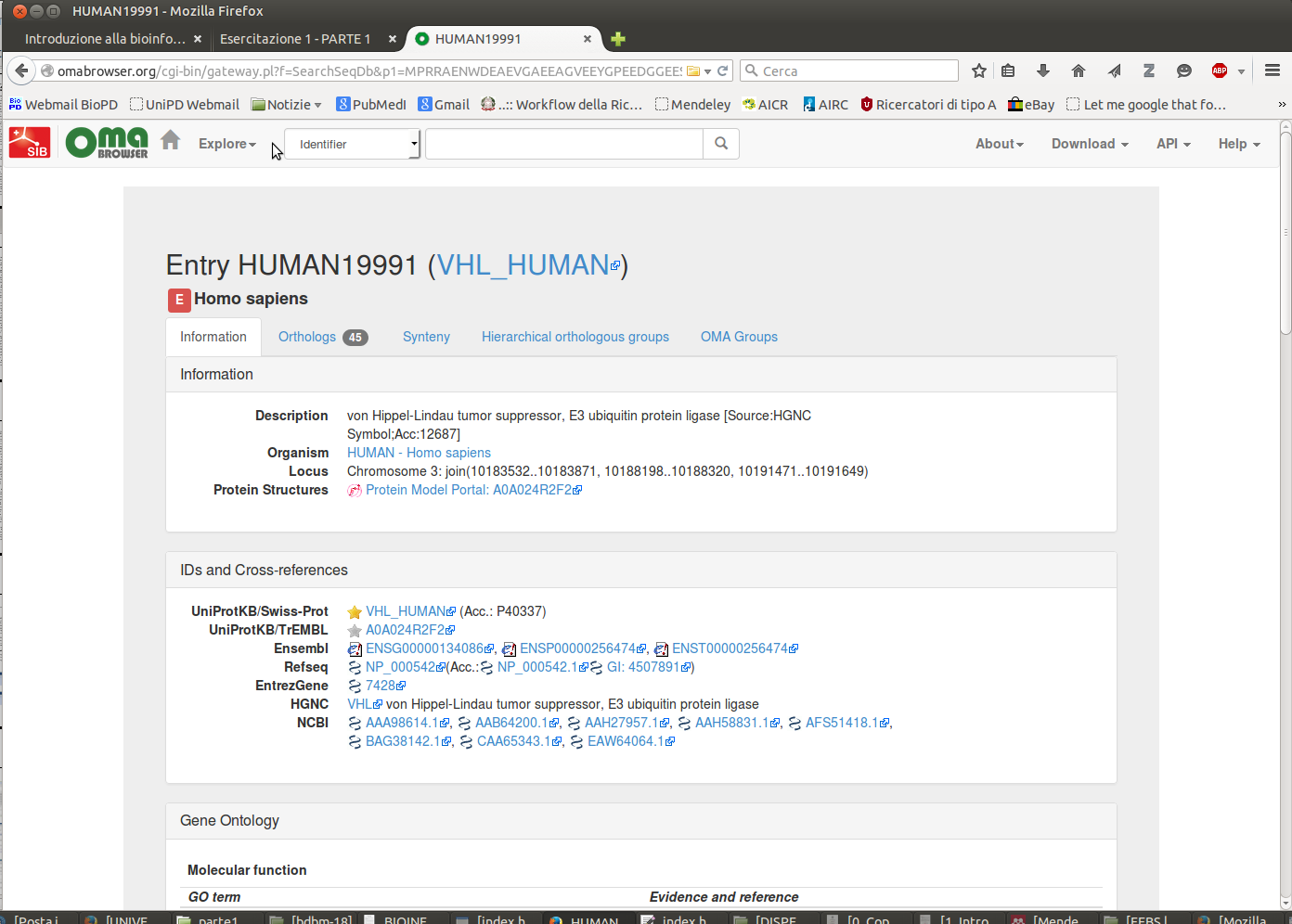
- Andate su OMA, selezionate nel box a sinistra Protein sequence ed inserite nel campo la sequenza amminoacidica della vostra proteina
- dovrebbe apparirvi qualcosa di simile a questo:
- Nella pagina vengono riportate informazioni relative alla proteina quali la descrizione generale e l'organismo di provenienza. Nel secondo box sono presenti inoltre dei link ad altri database esterni molto importanti per la nostra indagine.
- Selezioniamo ora il tab Orthologs . Ci verrà fornito un elenco con le proteine ortologhe individuate. In base alla proteina scelta, la lista può anche essere molto lunga o al contrario, contenere solo pochi ortologhi.
- Nella stessa pagina, in alto a destra, dovreste trovare un bottone Download (fasta) attraverso il quale scaricare le sequenze individuate. ATTENZIONE le sequenze verranno visualizzate come un file di teso (non è quindi un vero e proprio download), copiatele ed incollatele in un file di testo per poi salvarle... o più semplicemente usatele come input per il vostro programma di allineamento!
- Provate ora con la vostra sequenza
- Andate su Jalview e fate partire l'applicazione selezionando Launch Jalview Applet nel box in alto a destra
- In alternativa, potete utilizzare la versione locale di Jalview installata sul computer.
- In alternativa, potete scaricare il manuale ufficiale in formato PDF
- O una comoda presentazione introduttiva sul programma
- Analizziamo brevemente le informazioni contenute nella figura e proviamo a capire da dove vengono sulla base di quanto visto fino ad adesso.
- Provate voi a costruire una tabella riassuntiva con i risultati prodotti in questa esercitazione.
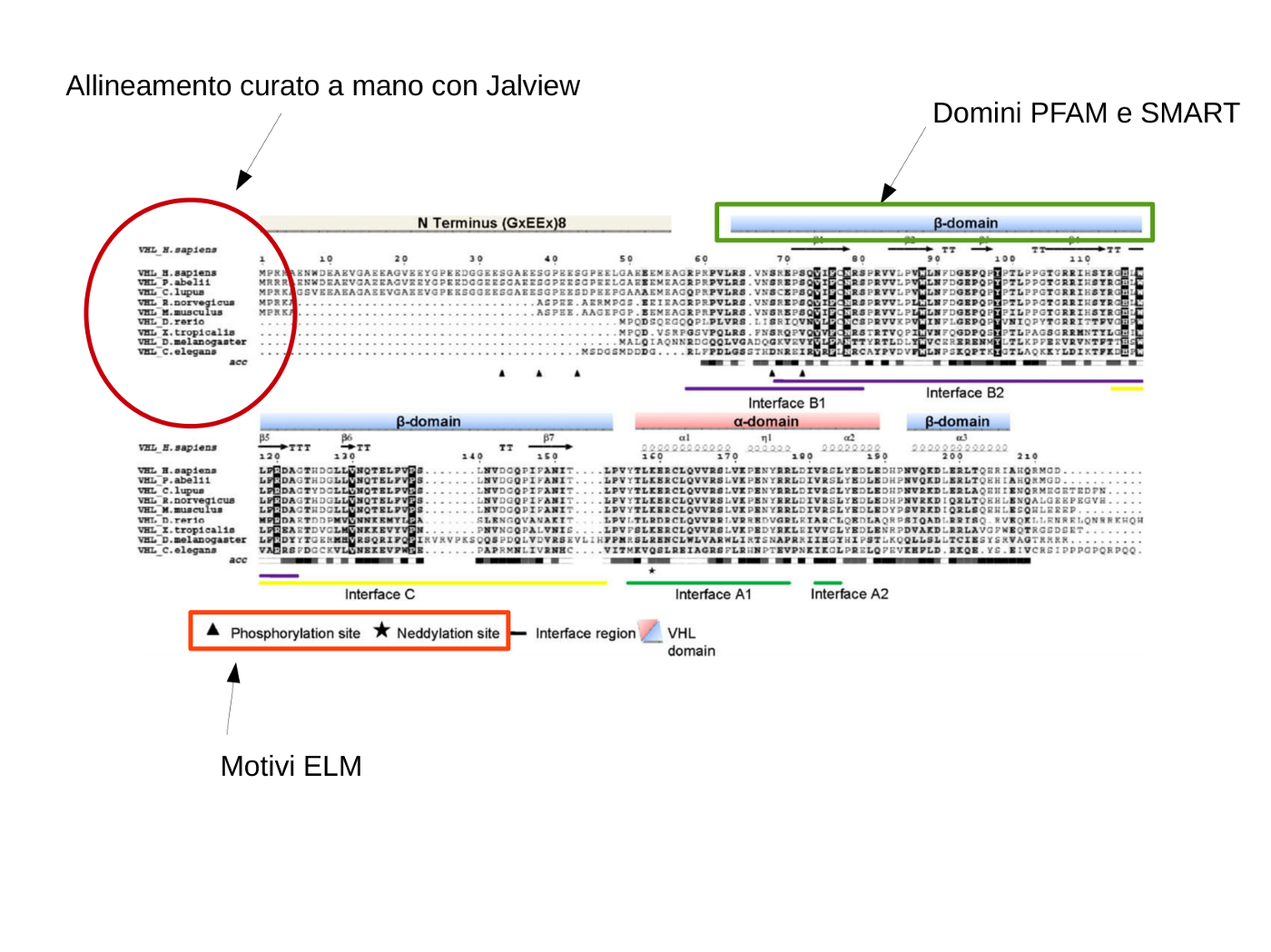
Che informazioni ottenete utilizzando le banche dati ed i servizi web elencati nella prima parte di questa esercitazione?
Per esempio, che tipo di informazioni ottengo interrogando i server
Questi due server in particolare risultano molto utili per individuare domini funzionali e motivi lineari. Tornando alla nostra proteina di esempio, interrogando il server PFAM otteniamo il seguente risultato
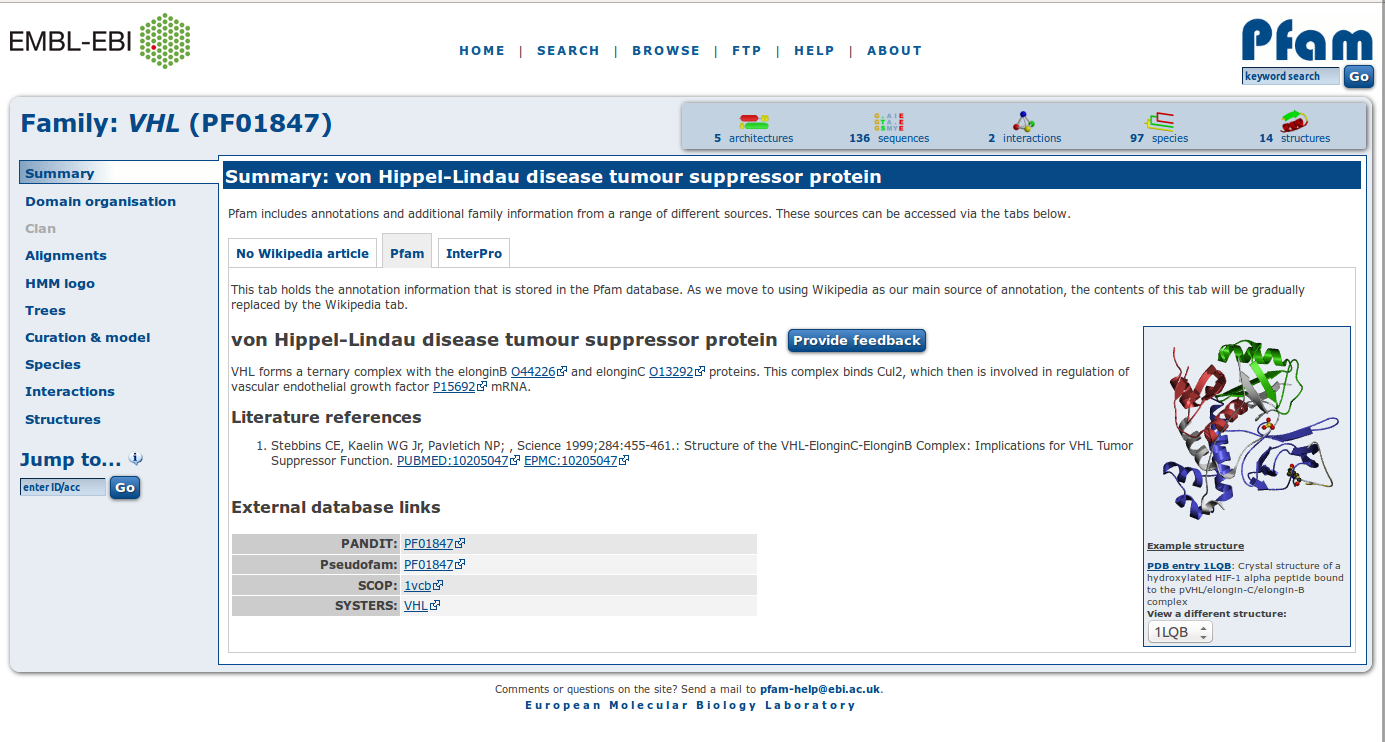
Il server identifica correttamente la nostra proteina evidenziando la presenza di un dominio definito "vhl".
Il server ELM ci è invece utile per identificare dei motivi funzionali in regioni non strutturate della proteina che prendono il nome di motivi lineari. A seguire un esemprio di ciò che ci viene fornito in output
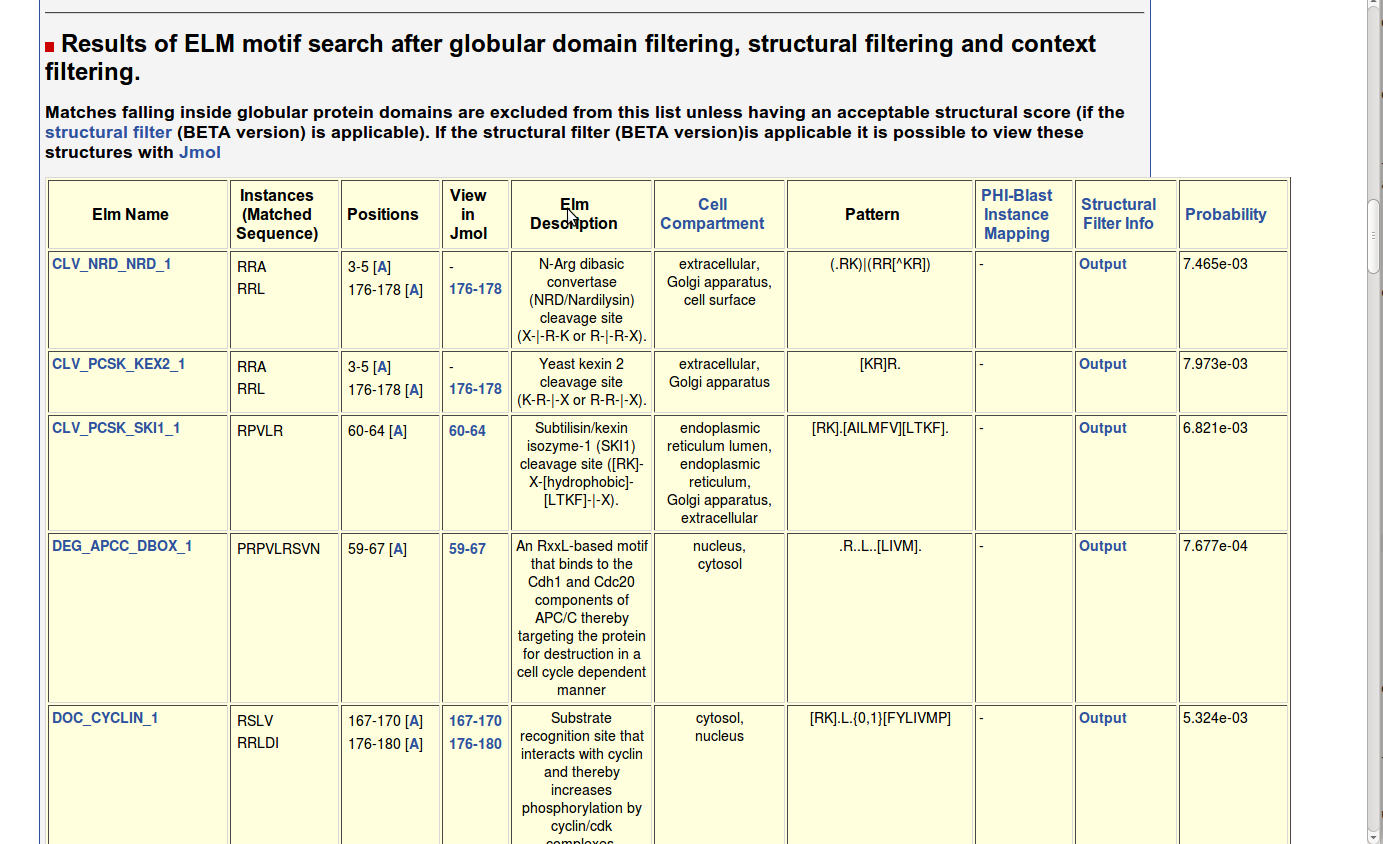
NOTA sono tutti affidabili i risultati proposti?
Allineare delle sequenze con Jalview
Un corretto allineamento rappresenta la base per una buona analisi bioinformatica. Seppur potenti, gli algoritmi di allineamento automatici non possono sostituire completamente l'operatore esperto. In altre parole, in questa fase dell'esercitazione vedremo come editare manualmente un allineamento di sequenza. Partiamo quindi con l'avviare Jalview
Un buon punto di partenza per l'utilizzo del programma ci è fornito dalla ricca documentazione disponibile in rete. Andate su Jalview_Documentation, nella sezione Getting Started troverete dei video introduttivi veramente ben fatti.
Allineiamo!
.... .... Inizio sessione interattiva ... ... Ci si aspetta arrivino domande!

Come organizzare i risultati finora ottenuti in maniera razionale
Finora abbiamo più che altro visto una carrellata di risorse bioinformatiche liberamente fruibili sul web. Abbiamo inoltre ottenuto parecchieindicazioni sulla nostra proteina ignota, ma al momento queste informazioni risultano più che altro confuse e non organizzate. Vedremo adesso come si possono organizzare i risultati ottenuti, come renderli presentabili e soprattutto, come fare in modo che le analisi condotte in questa sezione possano diventare del buon materiale per una pubblicazione.
Un esempio efficace di presentazione...
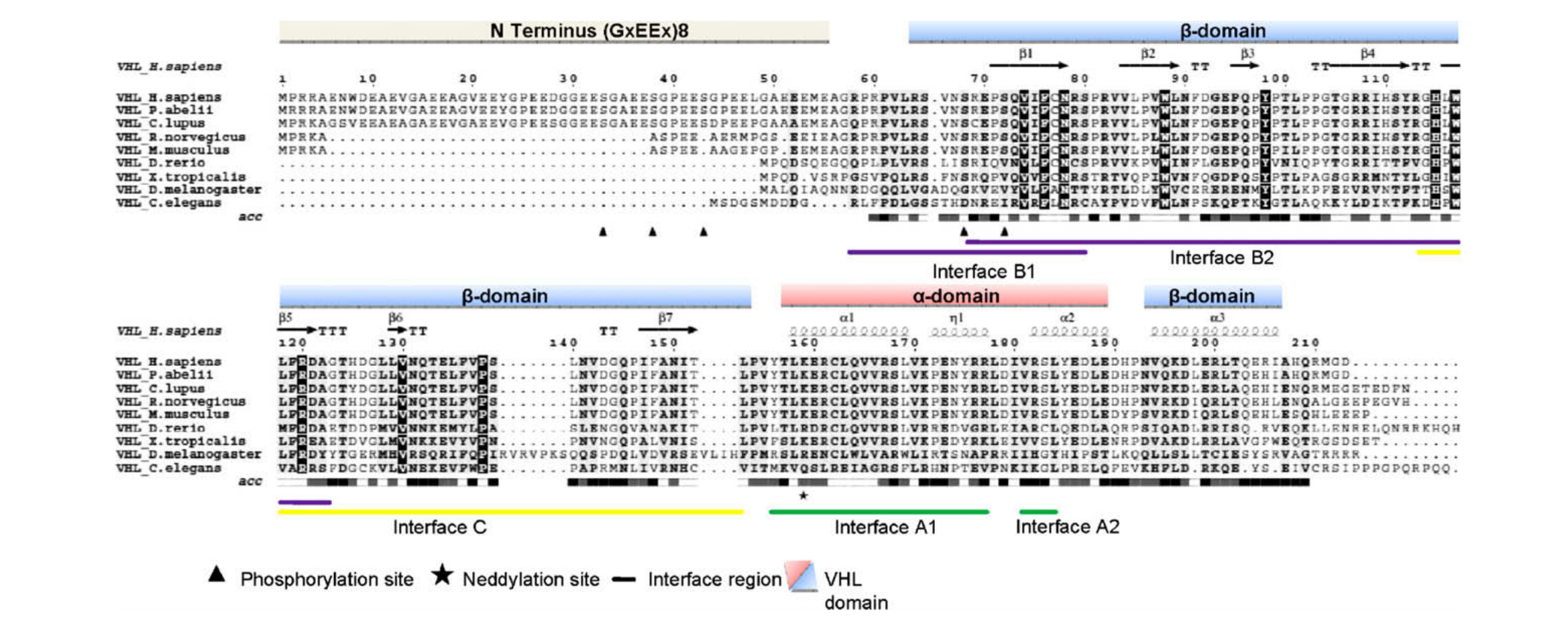
Scomponendo l'immagine nelle sue parti, otteniamo le seguenti informazioni: